Sampling the Protein - Backbone Moves
A combination of rigid body moves with internal moves are sufficient to sample most molecules. However, they will not allow you to efficiently sample the conformations of a protein. To understand why, let’s take a look at the script protein_angle.py, by typing
nano protein_angle.pyThis is a very similar script to angle.py, except this has been modified to change the angle between the C, CA and N atoms of random residues along the backbone of a protein. The lines;
## Set the maximum amount to change each angle
max_delta = 2.5 * degrees
## Set the temperature
temperature = 298.15 * kelvin
## Load the protein from the Sire restart file
system = Sire.Stream.load("protein.s3")specify the temperature, maximum amount to move each angle during the simulation, and then load the protein from the s3 file protein.s3. This is a copy of the protein that has been extracted from the l7n.top and l7n.crd Amber topology / coordinate files, using the script make_protein.py.
In the definition of the mcMove() function we have the code;
## get the version of the protein
## currently in the system
protein = system[ MolWithResID("ALA") ].molecule()This selects the protein from the system by finding a molecule that contains a residue called “ALA” (most proteins contain an alanine residue ;-)). Next,
## choose a random residue to move (will change the backbone angle)
res = ResIdx( rangen.randInt(0, protein.nResidues()-1) )
angleid = AngleID( res + AtomName("C"), res + AtomName("CA"), res + AtomName("N") )
print("\nRandomly chosen angle %s" % angleid)we have randomly chosen one of the residues in the protein by generating a random number between 0 and protein.nResidues()-1. The ResIdx object is a selector that selects the residue at that index in the protein. We then combine this residue selector with an AtomName selector, e.g. res + AtomName("C") would select the atom called “C” in the randomly chosen residue. We thus build an AngleID angle identifier that selects the C, CA and N atoms in that random residue. Next…
## randomly choose an amount to move the angle
delta = rangen.rand(-max_delta.to(degrees), max_delta.to(degrees)) * degrees
print("Randomly changing the angle by %s" % delta)
## change the angle
new_protein = protein.move().change(angleid, delta).commit()…we randomly change that angle in the protein using the .move().change() function as before.
Try running this script using;
$SIRE/bin/python protein_angle.pyThe output is the same format as the previous scripts and you can view the trajectory using
vmd output*.pdbBy eye, can you see if the acceptance ratio of these moves is high or low? Are most moves accepted, or are most moves rejects, or is it about 50/50?
What are the typical values for the change in energy for the move? Are any of these changes in energy greater than 30 kcal mol-1? What is the probability of accepting a move that has a change in energy of 30 kcal mol-1 at 298.15 K?
In the VMD movie, can you see where most of the moves are accepted? Are they located in the center of the protein, ends of the protein or at the hinge regions of the protein?
What you should see is that the majority of the moves are rejected. This is because moving an angle in the middle of the protein can result in large movements at the end of the protein (think of it like putting a small bend in the middle of a sheet of paper versus a small bend at the end of the paper - the bend in the middle will cause large changes at the ends). Moves in the middle cause big changes in the position of atoms nearer the ends of the protein, leading to steric clashes and thus large increases in energy. You should see that it was not uncommon to have seen increases in energy over 30 kcal mol-1 or even over 100 kcal mol-1 during the protein_angle.py simulation. If this is not clear, take a look at the protein backbone move section in this PDF file.
What this demonstrates is that internal bond, angle and dihedral moves cannot be used to sample the backbones of proteins. They can (and are) used to sample the bonds, angles and dihedrals in protein sidechains, but different types of move are needed to sample the backbone. There are several different algorithms (e.g. CONROT / concerted rotation), but the one used in Sire is perhaps the simplest. It is demonstrated in protein_backbone.py. The move works by recognising that the protein backbone can be viewed as a series of semi-rigid triangles formed by the C alpha (CA), O and N atoms of each residue, e.g. as shown here;
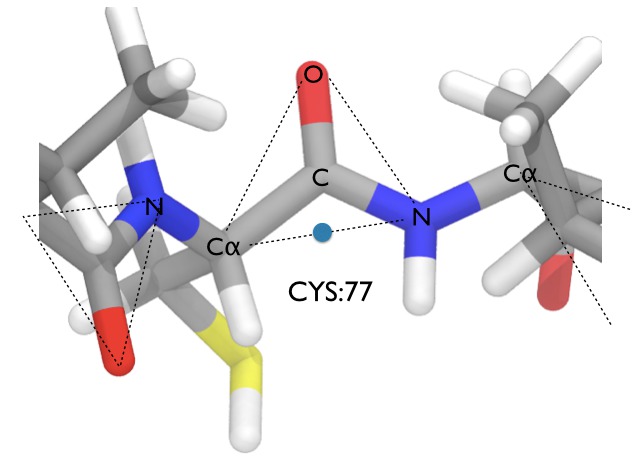
Because the bond between the CA and N atoms of neighbouring residues is quite soft, Monte Carlo can successfully translate each triangular unit as a single rigid body group. This means that we can use rigid body translations and rotations to sample each backbone triangle (with attached side-chain). This is performed in the script protein_backbone.py, which you can look at using
nano protein_backbone.pyThis is a slightly more complex script, but this complexity comes only from building the selection of atoms that comprise the backbone triangle plus sidechain. This is because the nitrogen in each triangle actually belongs to the next residue in the chain…
The code in mcMove() that performs the move is as follows;
## choose a random residue to move (will change the backbone angle)
res = ResIdx( rangen.randInt(0, protein.nResidues()-1) )
print("\nRandomly chosen residue %s" % res)
## Now choose a random vector by which to translate
## the residue
random_translate = rangen.vectorOnSphere( max_translation.value() )
## Now choose a random axis about which to
## rotate the residue molecule
random_rotate_axis = rangen.vectorOnSphere()
## Now choose a random amount by which to rotate
## the residue molecule
random_rotate_angle = rangen.rand(-max_rotation.value(),
max_rotation.value()) * degrees
print("Randomly translating by %s" % random_translate)
print("Randomly rotating by %s about %s" % \
(random_rotate_angle,random_rotate_axis))Here, we have first randomly chosen a residue to move, and then a random amount to translate and rotate that residue. Next we construct the selection of the backbone triangle plus sidechain atoms using
## Now select the residue, plus the neighbouring N atom
## (and deselect our N atom)
atoms = protein.select(res).selection()
N = protein.atom( res + AtomName("N") )
if res.value() < (protein.nResidues()-1):
try:
atoms.deselect( hn_atoms + res )
except:
pass
if res.value() > 0:
try:
atoms.select( hn_atoms + ResIdx(res.value()+1) )
N = protein.atom( ResIdx(res.value()-1) + AtomName("N") )
except:
passatoms contains the atoms that are selected. This first contains all of the atoms in the residue. We then deselect all of the N and HN atoms from this residue using atoms.deselect( hn_atoms + res ), where hn_atoms was defined at the start of the script to be;
hn_atoms = AtomName("N", CaseInsensitive) * AtomName("H", CaseInsensitive) * \
AtomName("HN", CaseInsensitive) * AtomName("HN1", CaseInsensitive) * \
AtomName("HN2", CaseInsensitive) * AtomName("HN3", CaseInsensitive)(these are the standard PDB atom names for the nitrogen and hydrogens bonded to that nitrogen).
We next select the N and HN atoms from the next residue in the chain using atoms.select( hn_atoms + ResIdx(res.value()+1) ). Note that this is all performed using python try: except: blocks to catch exceptions that are raised when we try to select the (non-existant) residue next to the last residue in the chain.
Next, we define the center of rotation of the move as the point that lies equidistant between the CA and N atoms (the blue point in the picture above) using;
## now get the center of rotation - this is the geometric
## center of the bond between the CA and N atoms
CA = protein.atom( res + AtomName("CA") )
center = CA.property("coordinates") + \
0.5*(N.property("coordinates")-CA.property("coordinates"))Finally(!) we can translate and rotate this selection of atoms using;
## now translate and rotate these atoms
new_protein = PartialMolecule(protein,atoms) \
.move() \
.rotate( \
Quaternion(random_rotate_angle,random_rotate_axis), \
center ) \
.translate(random_translate).commit().molecule()(PartialMolecule is a view of a molecule that can contain an arbitrary selection of atoms)
Try running this script by typing;
$SIRE/bin/python protein_backbone.pyThe output has the same format as before and you can visualise the trajectory by typing;
vmd output*.pdb- How does the acceptance ratio of these moves compare to
protein_angle.pyabove? - How does the change in energy associate with each move compare to
protein_angle.pyabove? Do you see many moves with changes in energy greater than 30 kcal mol-1? Or greater than 100 kcal mol-1? - Do you see much movement of the backbone in VMD?
You should see that more moves are accepted and the energy changes associated with each move have a much smaller magnitude than for protein_angle.py. However, you will also see that this (very short) simulation has not sampled the backbone well. You can view a video showing a very large number of backbone moves on this protein here.
As you can see in this video, this type of backbone move is good at sampling the local conformation of the backbone, but cannot sample concerted motions such as loop opening, hinge bending etc. Why do you think this is the case? How would you design a Monte Carlo move that could sample global motions of the protein backbone?